
Lopez Ramirez Lab
- About
- Contact
- People
- Publications
- Research
Dr. Lopez-Ramirez's Publications
We have been investigating the role of genes involved in Cerebral Cavernous Malformations (CCMs) diseaseand their implications in cardiovascular biology. CCMs are brain vascular malformations prone to repetitive hemorrhagic stroke that lead to significant morbidity and mortality in ~0.5% of the population. Currently, no pharmacologic therapy exists for CCMs. Thus, there is an urgent need to better understand CCM pathogenesis and identify therapeutic targets for CCM
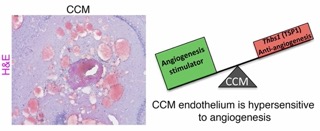 disease. We use bioinformatics, cell, and molecular techniques to analyze the effects of genetic inactivation of CCM genes in mouse brain microvascular endothelial cells (BMEC) and extended the results to humans and mouse models of the disease. Using these experimental approaches, we proposed that CCMs are hypersensitive to angiogenesis due to the loss of anti-angiogenic checkpoint protein thrombospondin 1 (TSP1). We found a profound CCM exacerbation as a result of Thbs1 (which encodes TSP1) loss of function mutations that also results in early lethality, establishing that the loss of TSP1 does indeed facilitate CCM formation (Lopez-Ramirez et al., Journal of Experimental Medicine, 2017).
disease. We use bioinformatics, cell, and molecular techniques to analyze the effects of genetic inactivation of CCM genes in mouse brain microvascular endothelial cells (BMEC) and extended the results to humans and mouse models of the disease. Using these experimental approaches, we proposed that CCMs are hypersensitive to angiogenesis due to the loss of anti-angiogenic checkpoint protein thrombospondin 1 (TSP1). We found a profound CCM exacerbation as a result of Thbs1 (which encodes TSP1) loss of function mutations that also results in early lethality, establishing that the loss of TSP1 does indeed facilitate CCM formation (Lopez-Ramirez et al., Journal of Experimental Medicine, 2017).

Our research has led to important work to understand the hemorrhagic stroke observed in CCM lesions. We discovered that during CCMs, there is a dramatic increase in factors that decrease blood coagulation (anticoagulant factors). These anticoagulant factors are notably low in normal young cerebral vasculature to prevent bleeding in the brain. Our observations led to the idea that the notable increase of the anticoagulant factors in the brain vasculature predisposes to cerebral hemorrhage during CCM disease. Indeed, We showed that hemorrhage in CCM is associated with locally elevated expression of the anticoagulant endothelial receptors thrombomodulin (TM) and the endothelial protein C receptor (EPCR) that generate activated protein C (APC) to form an anticoagulant vascular domain that might contribute to the bleeding-induced morbidity and mortality in CCMs (Lopez-Ramirez et al., Blood, 2019). Our current research aims to better understand the biological factors that enhance brain bleeding and ultimately aid in developing non-invasive therapies to prevent brain hemorrhages.
The propensity of CCM lesions to form in the central nervous system
parenchyma has never been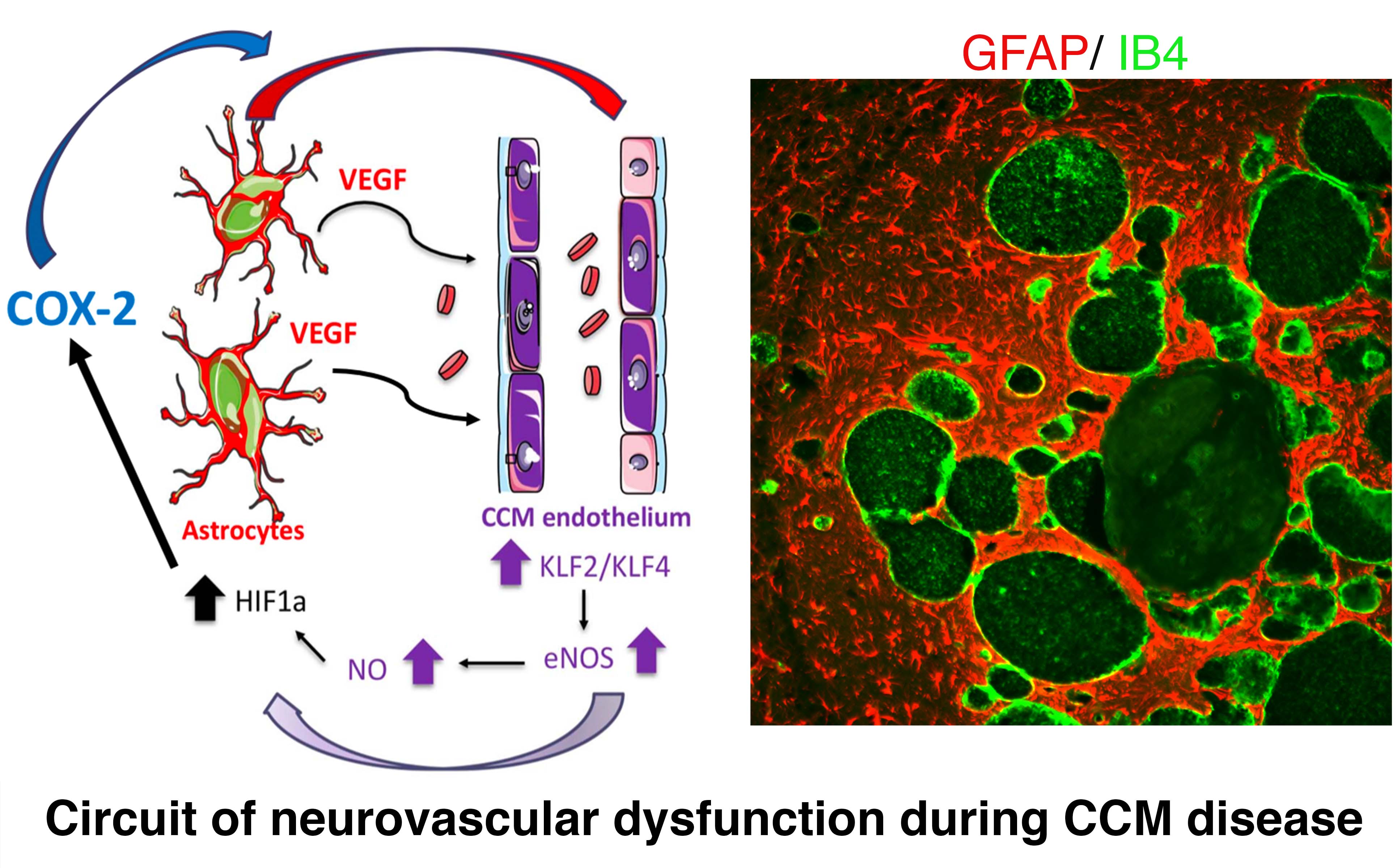 mechanistically clarified. We recently introduced a novel concept by which astrocytes make a significant contribution to cerebral cavernous malformations (CCMs) pathogenesis. We reported that proliferative astrocytes play a critical role in CCM pathogenesis by serving as a major source of VEGF during CCM lesion formation. An increase in astrocyte VEGF synthesis is driven by endothelial nitric oxide (NO) generated as a consequence of KLF2 and KLF4-dependent elevation of eNOS in CCM endothelium. Production of NO in CCM endothelium stabilizes HIF-1 in astrocytes, resulting in increased VEGF production and expression of a "hypoxic" program under normoxic conditions. We showed that pharmacological inhibition of HIF1-driven COX-2 by an approved and well-tolerated drug can ameliorate a murine model of CCM disease. Our study proposes a non-cell-autonomous mechanism mediated by CCM endothelium-driven hypoxia and angiogenic programs during disease. Our findings further support that components of the hypoxic program represent potential therapeutic targets for CCMs disease (Lopez-Ramirez et al., Journal of Clinical Investigation, 2021).
mechanistically clarified. We recently introduced a novel concept by which astrocytes make a significant contribution to cerebral cavernous malformations (CCMs) pathogenesis. We reported that proliferative astrocytes play a critical role in CCM pathogenesis by serving as a major source of VEGF during CCM lesion formation. An increase in astrocyte VEGF synthesis is driven by endothelial nitric oxide (NO) generated as a consequence of KLF2 and KLF4-dependent elevation of eNOS in CCM endothelium. Production of NO in CCM endothelium stabilizes HIF-1 in astrocytes, resulting in increased VEGF production and expression of a "hypoxic" program under normoxic conditions. We showed that pharmacological inhibition of HIF1-driven COX-2 by an approved and well-tolerated drug can ameliorate a murine model of CCM disease. Our study proposes a non-cell-autonomous mechanism mediated by CCM endothelium-driven hypoxia and angiogenic programs during disease. Our findings further support that components of the hypoxic program represent potential therapeutic targets for CCMs disease (Lopez-Ramirez et al., Journal of Clinical Investigation, 2021).

CCM lesion count, size, and aggressiveness vary widely among patients of similar ages with the same mutation or even within members of the same family. However, what determines the transition from quiescent lesions into mature and active (aggressive) CCM lesions is unknown. We observed that inflammation significantly contributes to CCM pathogenesis. We propose that inflammation and inflammasome activity play a critical role in transitioning from initial CCM formation into mature and active CCM lesions. Our findings further support that crosstalk between astrocytes and CCM endothelium can trigger the recruitment of inflammatory cells arising from brain parenchyma (microglia) and periphery immune system (leucocytes) into mature CCM lesions that propagate lesion growth, thrombosis, and bleeding (Chinhchu-Lai et al., BioRxiv, 2022). Our study reveals previously unknown links between neuroinflammatory astrocytes and inflamed CCM endothelium as contributors that trigger leukocyte recruitment and precipitate immunothrombosis in CCM lesions.

Vascular endothelial cells respond to hemodynamic forces from blood flow to exhibit vasodilatory, anticoagulant, fibrinolytic, and anti-inflammatory properties that confer vasoprotective effects. We found that genetic inactivation of endothelial Krit1 (Krev1 interaction trapped gene1, also known as CCM1) or Heg1 (Heart of glas1) leads to upregulation of Krüppel-like factors 4 and 2 (KLF4 and KLF2) expression, transcription factors central to the regulation of vascular homeostasis and flow-mediated vasoprotection. We also observed that vasoprotective proteins, such as endothelial nitric oxide synthase (eNOS) and thrombomodulin (TM), are upregulated by the increase of KLF2 and KLF4 in response to the loss of endothelial KRIT1. Moreover, the HEG1 binding pocket on the KRIT1 FERM domain is both discrete and unique, and structure-based modeling has identified a small molecule inhibitor with the capacity to disrupt the KRIT1-HEG1 interaction resulting in upregulation of endothelial KLF2 and KLF4 expression. We recently demonstrated that the small-molecule HEG1-KRIT1 inhibitor 2 (HKi2) is a bona fide inhibitor of the interaction between HEG1 and KRIT1 proteins by competing orthosterically with HEG1 through covalent reversible interactions with the FERM (4.1, ezrin, radixin, and moesin) domain of KRIT1. We showed that the crystal structure of HKi2 bound to KRIT1 FERM revealed that it occupies the same binding pocket on KRIT1 as the HEG1 cytoplasmic tail. Using human endothelial cells (ECs), acute inhibition of the HEG1-KRIT1 interaction by HKi2 increased KLF4 and KLF2 mRNA and protein levels, whereas a structurally similar inactive compound failed to do so. In zebrafish, HKi2 induced expression of klf2a in arterial and venous endothelium. Furthermore, genome-wide RNA transcriptome analysis of HKi2-treated ECs under static conditions revealed that in addition to elevating KLF4 and KLF2 expression, inhibition of the HEG1-KRIT1 inter-inhibition of the HEG1-KRIT1 interaction mimics many of the transcriptional effects of laminar blood flow and Krit1 KO endothelial cells (Lopez-Ramirez et al., 2021 FASEB bioadvances). We proposed that genetic or pharmacological disruption of the HEG1-KRIT1 interaction can mimic the vasoprotective effects of laminar flow on endothelium, thereby reducing thrombosis, atherosclerosis, and vascular inflammation.